Prionen sind Eiweißstoffe (Proteine), deren ungewöhnliche Eigenschaften die Biologen und Mediziner seit über 15 Jahren faszinieren. Offensichtlich sind Prionen die einzigen Eiweiße, die für ansteckende Krankheiten verantwortlich sind. Bis zur Entdeckung dieser ungewöhnlichen Partikel war man davon ausgegangen, dass alle Krankheitserreger auch Nukleinsäuren besitzen – jene fadenförmigen Moleküle, in denen die Erbinformation gespeichert wird.
Heute weiß man, dass mehrere Krankheiten des menschlichen Nervensystems, die durch geistigen Zerfall gekennzeichnet sind, auf das Konto der rätselhaften Prionen gehen. Zum Glück treten die langsamen, schleichenden Infektionen nur sehr selten auf: Etwa jeder millionste Mensch leidet unter der Creutzfeld- Jakob Krankheit (CJK), noch zehnmal seltener ist das Gerstmann-Sträussler-Syndrom.
Jahre, manchmal sogar Jahrzehnte können zwischen Infektion und Ausbruch der Krankheit vergehen; in dieser Zeit sind keinerlei Symptome festzustellen. Ist die Krankheit aber erst einmal ausgebrochen, verläuft sie unaufhaltsam und führt in der Regel zum Tod. Die Geschichte der Entdeckung der Prionen beginnt im Jahr 1956. Zwei amerikanische Forscher, Carleton Gajdusek und Vinzent Zigas, beobachten im Regenwald Neuguineas eine bis dahin unbekannte Seuche:
Kuru (aus der Sprache des Fore- Stammes für „zittern“) beginnt mit unkoordinierten, schüttelnden Bewegungen; es folgen Sprachverlust und Lähmungen, nach etwa einem Jahr tritt der Tod ein. Weit über tausend Patienten werden in den ersten Jahren untersucht. Für Gadjusek ist der Zusammenhang mit einer rituellen Form des Kannibalismus offensichtlich. Der Brauch, die toten Stammesangehörigen durch den Verzehr von Gehirnen zu ehren, ist mittlerweile erloschen. Speziell Frauen und Kinder nahmen an diesen Mahlzeiten teil, bei über 90 Prozent ist die Krankheit mittlerweile ausgebrochen. Wegen der langen Entwicklungszeit (diese kann über dreißig Jahre betragen) sind aber vereinzelt noch Fälle von Kuru zu beobachten.
Um die geheimnisvolle Krankheit besser untersuchen zu können, versuchten die Forscher jahrelang, den Erreger auf geeignete Versuchstiere zu übertragen. Als hilfreich erwies sich dabei eine Theorie von William Hadlow, der auf Gemeinsamkeiten zwischen Kuru und einer schon länger bekannten Tierseuche hinwies: Die Skrapie ist als Störung des Zentralen Nervensystems bei Schafen und Ziegen schon über 200 Jahre bekannt; das Krankheitsbild ähnelt sehr dem der Kuru.
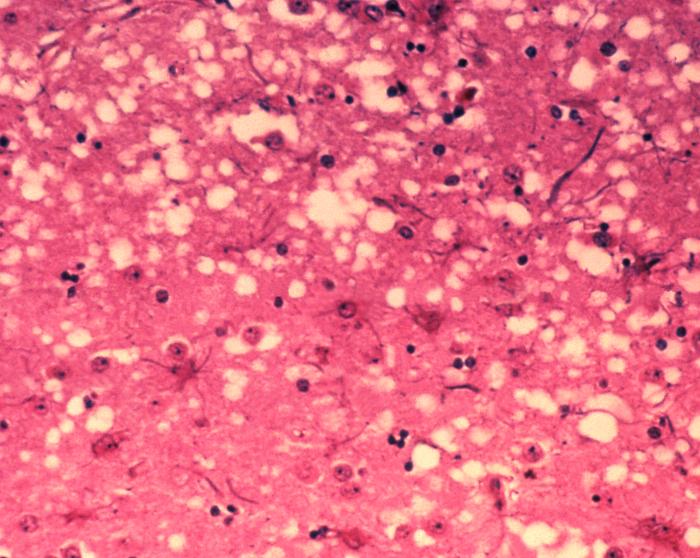
Auch die „Rinderseuche“ BSE wird von Prionen verursacht. Unter dem Mikroskop sieht man, dass die Infektion das Gehirn durchlöchert wie einen Schwamm. (Foto: Dr. Al Jenny via Wikimedia Commons)
Man wusste lediglich, dass sich die Prionen durch das Spritzen von Gehirngewebe zwischen den Tieren übertragen ließen. Um Näheres zu erfahren, mussten Gewebeproben aus dem Hirn infizierter Tiere in Fraktionen unterteilt werden, die sich in ihren physikalischen oder chemikalischen Eigenschaften unterschieden. Dann galt es herausfinden, in welcher Fraktion der Erreger am häufigsten vorkam – eine ungemein zeitraubende Aufgabe.
Bei den ersten Experimenten, die an Schafen und Ziegen durchgeführt wurden, musste eine ganze Herde jahrelang beobachtet und schließlich geopfert werden, um auch nur eine einzige Probe zu beurteilen. Erst 1978 gelang es Stanley Prusiner und seinen Mitarbeitern, das aufwendige Verfahren drastisch zu verkürzen. Mittlerweile kann die Aktivität einer Fraktion innerhalb von zwei Monaten an vier Hamstern ermittelt werden.
Gajdusek und seine Kollegen hatten inzwischen die Kuru auf Menschenaffen übertragen. Bald gelang dies auch mit der Creutzfeld-Jakob-Krankheit und dem Gerstmann-Sträussler-Syndrom. Die klinischen Merkmale und typischen Gewebeänderungen, die an den Versuchstieren festgestellt wurden, ließen auf eine enge Verwandtschaft der drei Krankheiten schließen.
Was dann im Laufe von Jahren an Fakten über die Prionen zusammengetragen wurde, versetzte die Fachleute in Erstaunen: Bis heute ist es nämlich nicht gelungen, bei den Partikeln Nukleinsäuren nachzuweisen. Behandelt man eine infektiöse Fraktion mit Biomolekülen, die diese Erbfäden zerstören können, bleiben die Skrapie-Erreger intakt. Auch UV-Bestrahlung in enorm hohen Dosen, Zink-Ionen und andere Chemikalien, die Nukleinsäuren zerstören, zeigen keine Wirkung auf die Prionen.
Da aber alle Lebewesen und sogar die Viren auf Nukleinsäuren angewiesen sind, um sich zu vermehren, entstanden allerlei Spekulationen über die merkwürdigen Krankheitserreger. Zeitweise gab es mehr Hypothesen über die Natur der Prionen als Arbeitsgruppen, die sich damit beschäftigten.
Die Hinweise, dass die krankmachenden Eigenschaften der Erreger auf ein Protein zurückzuführen sein könnten, mehrten sich. Proteasen (das sind Biomoleküle, die In der Lage sind, Eiweiße zu zersetzen) konnten die Ansteckungsfähigkeit der Skrapie-Erreger vermindern. Das gleiche gilt für andere Stoffe, die Eiweiße beschädigen. 1982 verkündete Prusiner schließlich seine Überzeugung, bei den Prionen handele es sich um ansteckende Eiweiße, und löste damit bei seinen Kollegen hitzige Diskussionen aus, die bis heute nicht verstummt sind.
Obwohl die Prionen selbst offensichtlich kein Erbmaterial enthalten, weiß man heute, dass die Informationen, die zur Herstellung dieser Proteine benötigt werden, in den Genen des Menschen und vieler anderer Säugetiere zu finden sind. Verwandte Proteine finden sich sogar bei Amphibien, Insekten und Hefen. Vermutlich spielt das Prion-Protein (PrP) eine wichtige Rolle für die Zelle, sonst wäre es im Verlauf der Evolution längst wieder verloren gegangen.
Alles deutet darauf hin, dass es mehrere Formen des PrP gibt. Während die „gesunde“ Form reibungslos funktioniert und eine noch unbekannte – für die Zelle wichtige – Aufgabe erfüllt, können „bösartige“ Varianten schwere Krankheiten verursachen. Die defekten PrP’s lagern sich nämlich zu fadenförmigen Gebilden (Fibrillen) zusammen, die sich im Gehirn absetzen. Der gleiche Vorgang ist auch bei der Alzheimerschen Krankheit und beim Down Syndrom zu beobachten, allerdings sind hier andere Proteine beteiligt.
Für das Gerstmann-Sträussler- Syndrom (GSS) hat Prusiner jetzt den Unterschied zwischen dem „gesundem“ und einem „kranken“ PrP herausfinden können. Bei dieser – erblichen – Krankheit genügt eine winzige Veränderung im Gen für das Prion- Protein. Dies hat zur Folge, dass ein einziger Baustein im Eiweiß vertauscht wird. Bei anderen Prion- Krankheiten – so spekuliert man – werden die schon fertigen Eiweiße in der Zelle nochmals verändert.
Die Frage, warum die Prionen alleine in der Lage sind, Kuru, CJK und GSS auszulösen, ist damit aber immer noch nicht geklärt. Dienen sie vielleicht als „Kondensationskerne“, an denen die Fibrillen entstehen? Oder greifen sie in die Verarbeitung der Erbinformation sein, so dass aus „gesunden“ Genen schadhafte Proteine entstehen? Auch nach über 30 Jahren intensiver Prionen- Forschung bleiben viele Rätsel ungelöst.
(erschienen am 2. Dezember 1989 in der WELT).
Was ist daraus geworden? Stanley Prusiner und Carleton Gajdusek erhielten beide den Nobelpreis, Prusiner erst acht Jahre nach diesem Artikel (1997), Gajdusek bereits 1976. Letzterer machte außerdem Schlagzeilen, weil er mehrere von ihm adoptierte Jungen aus Neuguinea und Mikronesien sexuell missbraucht hatte, und dafür auch zu einer Haftstrafe verurteilt wurde. Insgesamt hatte Gajdusek mit Einverständnis der Eltern 56 Kinder aus der Südsee in die USA geholt, die bei ihm aufwuchsen. Die Prionen erlangten ebenfalls traurige Berühmtheit als Verursacher der BSE-Epidemie. Der „Rinderwahn“ war 1986 in Großbritannien ausgebrochen, wo man das Fleisch mit Scrapie infizierter Schafe an die Kühe verfüttert hatte. Auf dem Höhepunkt der Epidemie erkrankten 1992 in Großbritannien 36000 Rinder, aber auch in Deutschland und in der Schweiz wurden insgesamt mehrere Hundert Fälle entdeckt.
Noch keine Kommentare